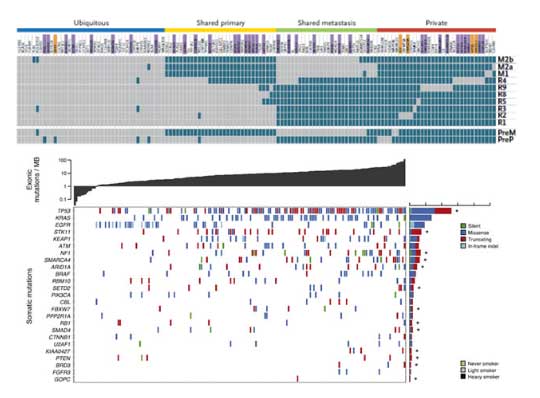
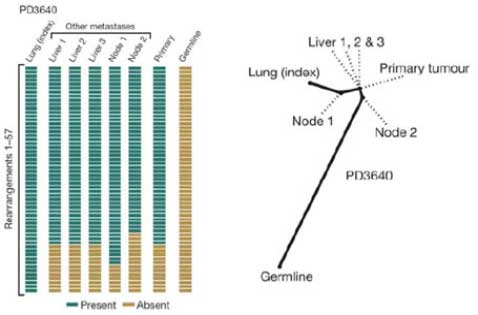
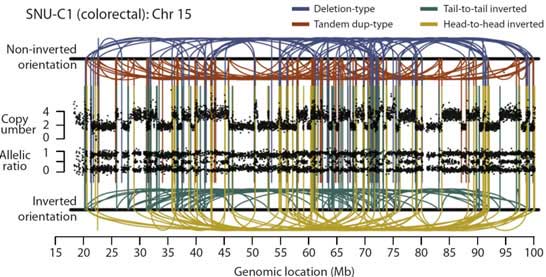
结构变异(StructureVariation,SV)是基因组变异的一类主要来源,主要由大片段序列(一般>1kb)的拷贝数变异(copynumbervariation,CNV)以及非平衡倒位(unbalanceinversion)事件构成。本分析模块将识别SNV结构及其断点(breakpoint),统计SNV类型在基因组内的分布状态,并与样本表性数据相关联。
SV形成机制包含:(1)同源性介导的直系同源序列区段重组(NAHR)(2)与DNA双链断裂修复或复制叉停顿修复相关的非同源重组(NHR)(3)通过扩展和压缩机制形成可变数量的串联重复序列(VNTR)(4)转座元件插入(一般主要是长/短间隔序列元件LINE/SINE或者伴随TEI相关事件的两者的组合)。
CRISPR-Cas9技术的应用已经在基础和临床研究、治疗、药物开发、农业和环境等领域得到了确认。临床研究显示,CRISPR在癌症、艾滋病、亨廷顿舞蹈症、杜氏肌肉营养不良症等疾病中具有潜在的应用价值。
CRISPR基因组编辑使研究人员能够快速、精准地创建转基因细胞系和动物模型。除了进行基因敲除和更具体的修饰外,研究人员还可使用CRISPR技术通过干扰(CRISPRi)或激活(CRISPRa)的方法调节基因表达,而不改变基因组序列。
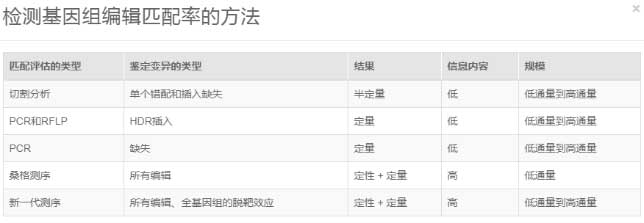
CRISPR基因组编辑实验得到的是混合细胞群,其中仅有一小部分细胞携带所需的编辑。研究人员需要确定哪些细胞具有所需的CRISPR敲除或靶向突变。目前评估编辑的方法包括切割分析、PCR、桑格测序和新一代测序(表)。
新一代测序是唯一一种在整个修饰范围内以高分辨率提供定性和定量信息的检测方法,能够满足任何通量的需求,并且可用于监测脱靶效应。7基于新一代测序的靶向测序通过关注靶向修饰区域,为确认CRISPR诱导的编辑提供了一种经济有效的解决方案。
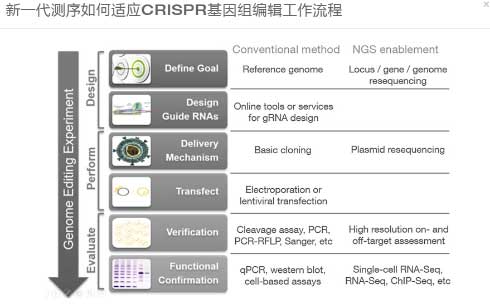
除了高分辨率的匹配和脱靶评估、功能验证以及对CRISPR敲除和编辑的评估外,新一代测序还可以在CRISPR基因组编辑工作流程的其他阶段整合。
在最初的设计阶段,一个基因位点或基因组的重测序(对于缺乏参考基因组的物种)有助于RNA选择。在克隆CRISPR-Cas9/向导RNA构建的过程中,对得到的质粒进行重测序,可以快速、高度可信地验证CRISPR的递送载体,特别是对于有大型质粒文库的高通量实验。
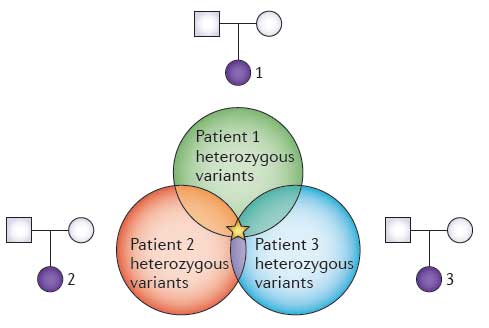
对于单基因致病位点,可以对有近亲关系和非近亲血缘关系的个体通过捕获并测序外显子区域上的各种类型的突变情况,包括SNP和 InDel,进行筛选。根据外显子组测序结果并结合假设的遗传模型准确地推断疾病位点,我们可以对于纯合型或杂合型的SNP以及indel进行系统详细的 功能性分析,包括蛋白突变体危害性评估,突变体蛋白结构解析,最终阐释单基因病因与分子变异的功能的关系。同时,利用自有开发算法可以对近亲婚配子女或非 近亲婚配子女患病的共享的潜在的疾病易感位点、片段完成推断问题;或是来自Trios 设计的样本寻找患者特有的de novo 突变。
对于染色体疾病,一般可以采用CNV芯片或(低覆盖度)全基因组测序分析解析染色体疾病致病区域。
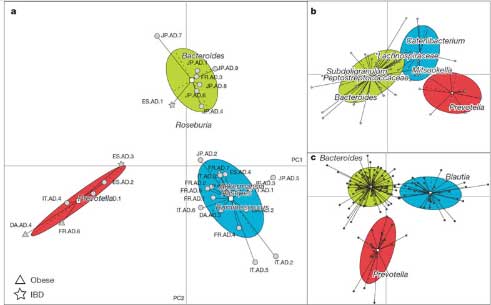
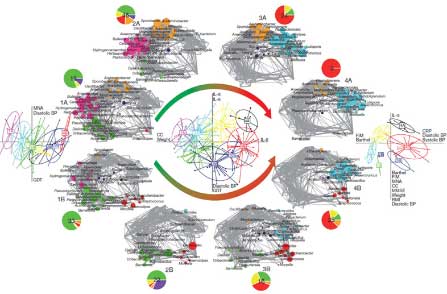
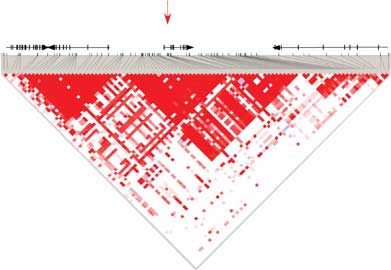
GWAS全基因组关联分析的主要目的是识别疾病关联的SNP位点和CNV区域,通过具有LD-关联连锁关系的SNP标记来捕获潜在的易感基 因位点。在大部分GWAS研究中,我们通过tagSNP(标签SNP)来推断预测和疾病关联的位点位于何处。后续GWAS分析将针对SNP疾病易感位点进 行评估和功能验证。
根据研究设计不同和研究表型的不同,采用的统计分析方法亦不同。如病例对照研究设计(质量性状),比较每个SNP的等位基因频率在病例和对 照组中的差别可采用4格表的卡方检验,计算相对危险度(Odds Ratio,OR值)及其95%的可信限。此外,还需要调整主要的混杂因素,如年龄、性 别等。这里采用logistic回归分析,以研究对象患病状态为因变量,以基因型和混杂因素作为自变量进行分析。我们也将针对对全基因组SNP位点进行关 联分析采用 additive模型Cochran-Armitage trend test ; 全基因关联结果的Q-Q plot , 可以评估是否存 在系统性的偏差。关联分析的结果可视化展示,根据LD连锁关系将多SNP与表型性状进行关联展示(如下图所示)。







高通量测序数据日益加大,呈指数型积累上升,必须交由高速网络提速分析。
基于以色列Infiniband技术,惠研云构建优化了集群计算性能,使得计算IO提升300%,持续数据交互速率提高到2T总带宽,保证集群中任务与数据0延迟,无阻塞。

长按识别或截图保存
关注惠研生物公众号
 公司介绍
公司介绍 发表文章
发表文章 新闻与活动
新闻与活动 招贤纳士
招贤纳士