

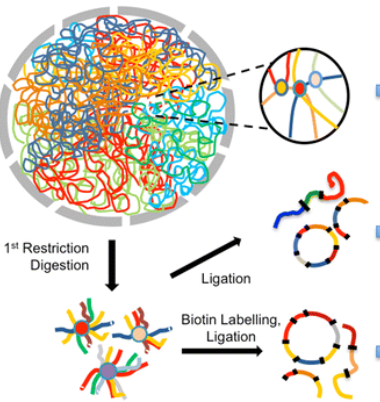

外显子组测序可能是应用最广泛的靶向测序方法。外显子组(人类基因组中的蛋白编码区域)只占基因组的2%,但它包含了约85%已知与疾病相关的变异,这使得全外显子组测序成为全基因组测序经济高效的替代方法。外显子组测序利用外显子组富集技术可有效识别包括群体遗传学、遗传疾病和癌症研究等一系列广泛应用中的编码变异。
优势: 在多种应用中识别变异 实现编码区域的全面覆盖 提供了一种经济的全基因组测序替代方法(每个外显子组只需检测4–5 Gb数据,而每个人类全基因组需检测约90 Gb数据)与全基因组测序相比,数据集更小、更易于管理,可以更快、更容易地进行分析

16S核糖体RNA (rRNA) 测序是一种常用的扩增子测序方法,用于鉴定和比较特定样品中存在的细菌。16S rRNA基因测序作为一种成熟的方法,适用于研究复杂微生物组或环境中样品的系统发育和分类,而这些研究之前很难或无法开展。16S研究的数据可改善分类指定的灵敏度和特异性,使其下降到物种的水平。
与毛细管电泳测序或PCR方法不同,新一代测序 (NGS) 是一种无须培养的方法,可实现样品中整个微生物种群的分析,包括鉴定那些利用其他方法未发现的物种。通过在一个测序运行中集合多个样品的能力,微生物学研究人员能够经济高效地开展基于NGS的16S rRNA测序,以鉴定那些利用其他方法未发现的菌株。


长按识别或截图保存
关注惠研生物公众号
 公司介绍
公司介绍 发表文章
发表文章 新闻与活动
新闻与活动 招贤纳士
招贤纳士

